bat365正版唯一官网
杨吉春课题组在糖尿病及血管病变发病机制研究中取得系列重要进展
糖尿病已成为严重危害我国国民健康的重大疾病,急需对其发病机制展开更深入研究以寻找新的干预策略。胰岛功能障碍和胰岛素抵抗是2型糖尿病发生的核心环节。杨吉春教授课题组的前期研究揭示FAM3A 是一个新线粒体蛋白,其促进ATP合成并抑制ROS生成。FAM3A诱导释放的ATP作用于细胞膜上的P2受体,进而以非胰岛素依赖途径激活Ca2+-CaM (Calmodulin)-PI3K-Akt通路抑制肝脏糖异生。FAM3A还激活AMPK通路,抑制脂质合成并增加肝脏脂质氧化。肥胖时,转录因子NFE2被游离脂肪酸激活后,通过诱导miR-423-5p表达进而抑制FAM3A表达,导致高血糖及肝脏脂质沉积。此外,FAM3A是核受体PPARγ的一个新靶基因,其介入了PPARg对糖脂代谢的调控过程。ATP合酶β亚基(ATPSβ)是线粒体ATP合酶的催化亚基,糖尿病发生时,肝脏ATPSβ表达下降与ATP含量下降及糖脂代谢异常相关。在肥胖小鼠肝脏过表达ATPSβ能提高线粒体质子回流速率、提高ATP合成、降低膜电位抑制ROS生成,从而缓解糖尿病和脂肪肝。FAM3A促进ATP合成与上调ATPSβ表达有关(Diabetes2017;Hepatology2014;Diabetes 2014; Biochim Biophys Acta 2013)。
FAM3A也高表达于人及动物胰岛β细胞中,且在糖尿病发生时显著降低。特异敲除β细胞FAM3A基因后,小鼠出现严重的胰岛素合成分泌障碍。在胰岛β细胞中,FAM3A-ATP-P2受体通路促使CaM入核,作为FOXA2的转录共激活因子,诱导PDX1及胰岛素基因表达。肥胖时,FAM3A被miR-423-5p抑制,导致胰岛素合成分泌障碍。(FASEB J. 2020 Mar;34(3):3915-3931.)。
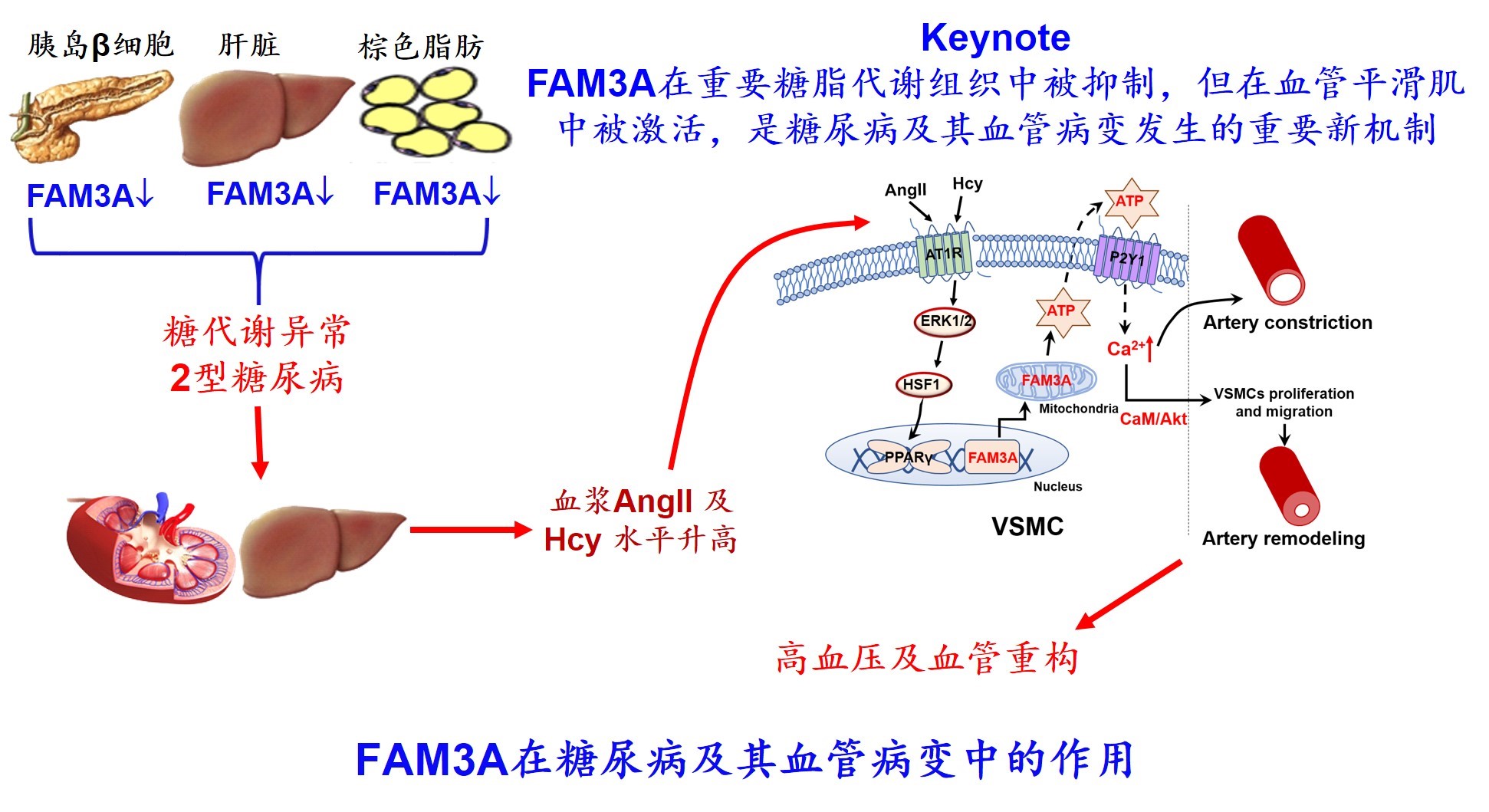
FAM3A也表达于血管平滑肌细胞中,其在促进血管平滑肌细胞增殖及血压调节中也起重要作用。肥胖时,重要糖代谢组织中FAM3A表达被抑制,促使血糖升高。高血糖导致血浆血管紧张素II(AngII)及同型半胱氨酸(Hcy)水平升高,其在血管平滑肌细胞中通过激活HSF1-PPARg信号轴诱导FAM3A表达,刺激细胞由收缩表型向增殖表型转化,从而导致高血压及心血管重构。在高血压患者及动物的动脉平滑肌层中,HSF1-PPARg-FAM3A信号轴被显著激活。FAM3A-ATP-P2受体通路在糖代谢组织中被抑制,但在血管平滑肌中被激活,是糖尿病血管病变发生的重要新机制(Circulation Research. 2020 Apr 13. doi: 10.1161/CIRCRESAHA.119.315558. [Epub ahead of print])。
很显然,激活代谢组织中的FAM3A表达,是糖尿病及其并发症治疗的潜在干预策略。药物筛选发现,抗抑郁药多虑平在肥胖小鼠肝脏及棕色脂肪组织中通过激活转录因子HNF4a诱导FAM3A表达,抑制肝脏糖异生与脂质合成并增加棕色脂肪产热,从而缓解高血糖、脂肪肝及肥胖症状。多虑平对白色脂肪组织及心血管系统中的FAM3A表达没有影响。多虑平对代谢异常的改善作用在敲除FAM3A基因后丧失。显然,激活糖脂代谢组织中FAM3A表达是糖尿病及脂肪肝的有效干预手段。此外,由于糖尿病患者是抑郁症的高危人群,本研究为优先推荐多虑平作为糖尿病合并抑郁症患者的抗抑郁药提供了理论基础(Diabetes2020 ,https://diabetes.diabetesjournals.org/content/early/2020/04/17/db19-1038)
上述课题受到国家自然基金、科技部慢病项目和精准医学项目及北京市重点项目的资助。杨吉春是上述系列研究论文的通讯作者,第一作者为课题组研究生。